
For the past week I’ve been working on a small but important facet of my inverse modeling research: a sensitivity analysis. Since some of the constraints placed on the model are estimated from measured quantities and not really nailed down, we need to be confident that a small change in the measurement wouldn’t make a large change to the results. To do this, we simply have to run the model over again with a new, estimated value. For example is we said that Quantity 1 = 100 originally, we could try Quantity 1 = 120 or 80 and see how the model responds. Hopefully that all sounds pretty easy because it is, the hard part is interpreting the results in a (somewhat) rigorous way.
Before getting too much further I need to confess that I am NOT a statistician at all. In fact, the only formal education I have in stats is the highschool material that I taught myself for the AP test. It will be quite obvious that I am well out of my league here, but I’ve come to believe that statistics is a field that’s out of everyone’s league[1].
The results of my model consist of 1,000 vectors of 105 components with each component representing a nutrient flow in the ecosystem. Each of these 1,000 vectors is sampled via a Markov Chain Monte Carlo simulation, which is simply a random walk procedure. Although I am still in the process of proving this, it can be assumed that each of these vectors are independent and normally distributed. So far I have always used the derived statistics (mean and SD) of this 1,000 element distribution to compare different model runs, but I now realize there is no simple t-test that can tell me if the sensativity runs are significantly different.
\[ X_1 = \left( \vec{x_1}, \vec{x_2}, \vec{x_3}, \cdots \vec{x_{1000}}\right) = \left( \begin{array}{c} x_{1,1},x_{2,1}, x_{3,1} \cdots x_{1000,1} \\ x_{1,2},x_{2,2},x_{3,2} \cdots x_{1000,2} \\ x_{1,3},x_{2,3},x_{3,3} \cdots x_{1000,3} \\ \vdots \\x_{1,105} \cdots \cdots \cdots x_{1000,n} \end{array} \right) \\\text{ compared to } X_2 = \left( \vec{x_1}, \vec{x_2}, \vec{x_3}, \cdots \vec{x_{1000}}\right) = \left( \begin{array}{c} x_{1,1},x_{2,1}, x_{3,1} \cdots x_{1000,1} \\ x_{1,2},x_{2,2},x_{3,2} \cdots x_{1000,2} \\ x_{1,3},x_{2,3},x_{3,3} \cdots x_{1000,3} \\ \vdots \\x_{1,105} \cdots \cdots \cdots x_{1000,n} \end{array} \right)
\]
I spent a few days reading up on statistical tests with particular emphasis on non-parametric tests and multivariate analysis in the hopes of finding a good avenue for analyzing my model. Nevertheless the answer–so far as I’ve found one–came to me after countless hours of struggling and had nothing to do with my search. In fact as it so often happens, the idea came to me suddenly and was based off some previous work I did a few years ago into machine learning. One aspect of machine learning that I had found particularly interesting was the idea of Self Organizing Maps (SOM) whereby novel information can be input to the computer and then projected into lower dimensional space while retaining the topological relationships of the original (see my original notes here). SOM is related to clustering and that was just what I needed to analyze my model runs.
Since clustering is a huge field with many books dedicated to each little part of it, I will leave substantive discussion of the topic to another time. Here I will simply show off some of the progress that’s been made.
The first two methods that we’ll look at attempt to fit the dataset into however many groups you expect to find. While this may not seem like an ideal place to start an analysis (what if the number of groups is unknown?), the methods are simple and intuitive. From there we’ll look at a more intriguing, yet risky, method.
PAM
Partitioning Around Medoids, otherwise known as PAM, is a well-behaved and self contained procedure that iteratively determines the most central data points as the basis for the groups. Okay, so what does that mean? Let’s start with an example in 2D.
## Let's first take a look at the data
plot(data,main="Dataset", xlim=c(0,10), ylim=c(0,10), pch=7)
## Using cluster::pam we will ask it for 2 clusters
require(cluster)
pam.data = pam(data,2)
##Let's take a look at the results
color= c("purple", "blue")
plot(NULL, xlim=c(0,10), ylim=c(0,10), xlab="x", ylab="y", main="Clusters")
points(data, col=color[pam.data$clustering], pch=7)
points(pam.data$med, col='red', pch=15)
plot(pam.data) ## pam can generate it's own plots too.
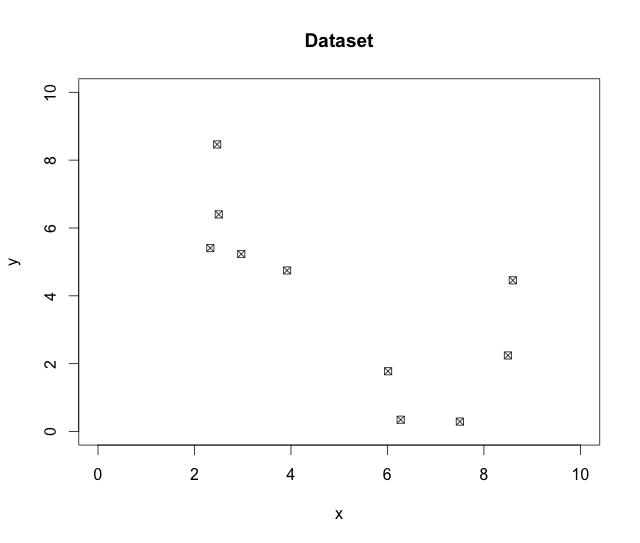
As seen in Figure 1, there is definitely some evidence of two groups in the data. Let’s see what PAM picks out of this.
In Figure 2 I’ve color coded the two clusters with purple and blue, respectively, along with the two Medoids in red. Visually this assignment looks quite accurate, but the choice of the medoid in the second groups seems a bit arbitrary. We’ll look into that right after we skim over how the algorithm works.
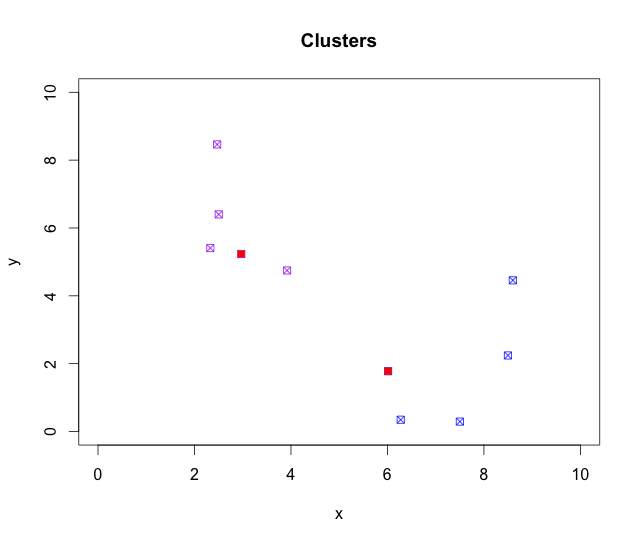
The way PAM works is through an iterative process of picking medoids and calculating scores based on distance. First the algorithm picks medoids from random, so in our case 2 points from the 10 in the data. It then groups the remaining points based on which points are closest and not yet assigned a group–much like captains picking teams for kickball. The total distance between the medoid and it’s group members is then calculated, and if a different member of the group offers a better score the medoids switches. Any switch brings us back to the beginning and new groups are picked based on the new medoids. This process continues until no better medoids are found.
One of the interesting aspects to this clustering methods involves the fact that the medoid is always an actual point. In fact that is where the term medoid comes from: median.
On aspect that we won’t cover here, but which almost all clustering algorithms do is Principal Coordinate Analysis. PCA is a technique to rotate and scale the data so that the variance within the data is best represented graphically. While nothing more than a way of representing data, it’s important to touch upon since the actual PAM plot will look different from the original because of it.

K-means
Now that we’ve done most of the drudge work, the explanation of k-means will be quick and simple. For k-means, the algorithm works quite similarly to PAM except instead of using data-points as medoids, it makes new points that are called ‘centers‘.
km.data = kmeans(data, 2) plot(data, col=color[km.data$cluster], pch=4, xlim=c(0,10), ylim=c(0,10), main="k-means") points(km.data$centers, pch=15)

Notice that along with our familiar groups there are two more points (black). These are the centers of the two clusters and were chosen in order to minimize the distance between them and the points in the cluster.
Hierarchical Clustering
The next class of clustering techniques is based on the distance between each of the data points. Much like some of those old tables where the distances between cities were listed as a large chart, here it is represented as a matrix:
> dist(data)
1 2 3 4 5 6 7 8 9
2 3.9883355
3 1.0665399 3.2687058
4 1.7228049 3.0573977 0.6648023
5 2.1759735 2.0631174 1.2568682 1.0066989
6 5.7170699 9.5959660 6.7047194 7.2765157 7.8923028
7 5.2201176 8.6628960 6.2863111 6.9356026 7.2957548 2.1896458
8 3.6338843 7.5664837 4.6046946 5.1734897 5.8038544 2.1030509 2.5288978
9 4.6904881 7.3232090 5.6843016 6.3439676 6.3987436 4.3073031 2.2175264 3.7253355
10 4.9916397 8.9640418 5.8993701 6.4191497 7.1322142 1.2275771 2.9210781 1.4542227 4.7221373
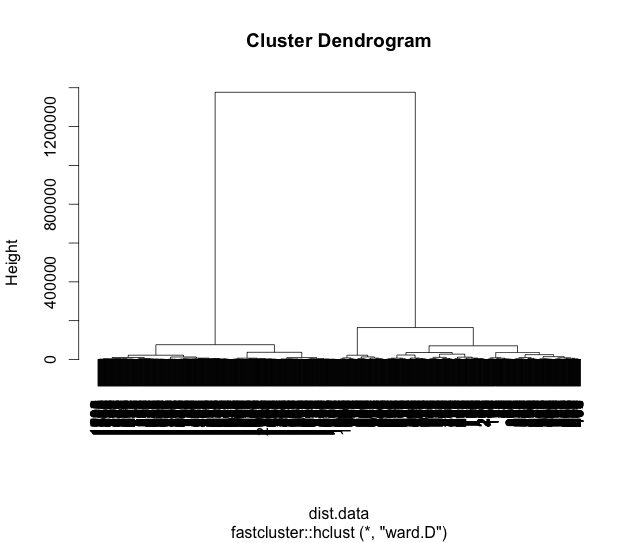
Typically in hierarchical clustering the two closest points are merged and then the process is repeated using their average distances. This process leads itself well to graphical representation as a dendrogram (from the Greek δενδρον meaning ‘tree’).
require(fastcluster) hc.data = fastcluster::hclust(dist(data), "ward.D") plot(hc.data)

Once again our clusters have remained intact as seen by the lengthy arms of the tree linking the left to the right hand sides. We can now see, among other things, that points 3 and 4 are more similar to each other than to point 5. In fact, a dendrogram is a terrific way to see many of the relationships between the data, but it can also lead to false conclusions since the difference between two points might look large when in fact there may be very little. Like anything, it’s a trade off.
Now that we have surveyed three techniques here, let me simply show the output when comparing two runs of my model.

See my notebook at http://misc.tkelly.org/R/Statistics.html
- Considering there are very few facts in statistics. The frequentists say that Bayesians make up everything, and the Bayesians think that the frequentists just sit around counting stuff and making big claims to significance.
2 thoughts on “Statistical Analysis for my Inverse Modeling”
Comments are closed.